
FDA’s Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use


The FDA issued guidance in November 2017 in connection with a comprehensive policy framework for the development and oversight of regenerative medicine products, including human cell and tissue products (HCT/P). Based on that guidance, the FDA determined that amniotic fluid is regulated as a Section 351 Biologic and requires an approved Biologics License Application (BLA) for it to be commercialized. In their 2017 guidance, the FDA allowed a 36-month grace period during which the FDA would exercise enforcement discretion. The enforcement discretion was extended last year due to COVID, but will end May 31, 2021. Effective June 1, 2021, all amniotic fluids are considered Section 351 Biologics. Absent a biologics license, no amniotic fluid can be commercialized. In order to ensure compliance with these changes, Russell Health will cease selling and marketing of amniotic fluid effective Monday May 24, 2021.
Since 2019, the FDA has issued more than 350 Warning Letters and Untitled Letter to manufacturers, clinics and health care providers related to unapproved regenerative medicine products. According to Dr. Peter Marks, Director, Center for Biologics Evaluation and Research, the FDA will not extend the November 2017 Regenerative Medicine Policy Framework, reaffirming the end of the compliance and enforcement discretion policy for certain human cell, tissue and cellular and tissue-based products (HCT/Ps), including regenerative medicine therapies. The period during which the FDA intends to exercise enforcement discretion with respect to the IND and premarket approval requirements for certain HCT/Ps ends on May 31, 2021, and will not be extended further. FDA slightly updated the 2017 guidance in July 2020: Regulatory Considerations for Human Cells, Tissues, and Cellular and Tissue-Based Products: Minimal Manipulation and Homologous Use; Guidance for Industry and Food and Drug Administration Staff, (fda.gov).
Despite the FDA’s effort to participate with the industry, there continue to be broad marketing of unapproved products for treatments or cure of a wide range of diseases or medical conditions. It is imperative that HCT/P manufacturers, as well as stakeholders that market HCT/Ps – including distributors, billing companies and consultants, health economic consultants, clinics and health care providers – assure they are in compliance with FDA standards. Furthermore, CMS has communicated to manufacturers and distributors that they expect a letter from FDA’s Office of Combination Products or FDA’s Tissue Reference Group in order to establish reimbursement as a section 361 product.
While Russell Health will no longer be distributing acellular amniotic fluid products, we will continue to distribute our 361 Human Cellular and Tissue-based product lines. Human Tissue Therapy products attempt to utilize acellular, minimally manipulated tissue allografts and are comprised of tissue allograft components intended for homologous use to supplement tissue. Tissue allografts are classified by the Food and Drug Administration (FDA) as Human Cell, Tissue and Cellular and Tissue-Based Products (HCT/P) that are regulated solely under section 361 of the Public Health Service (PHS) Act. FDA recognizes that human tissue was designed, or evolved, to perform certain functions in the human body with exquisite safety and effectiveness. As an HCT/P regulated solely under the Section 361 of the PHS Act, tissue allografts are exempt from FDA pre-market review, clearance, and approval from FDA.
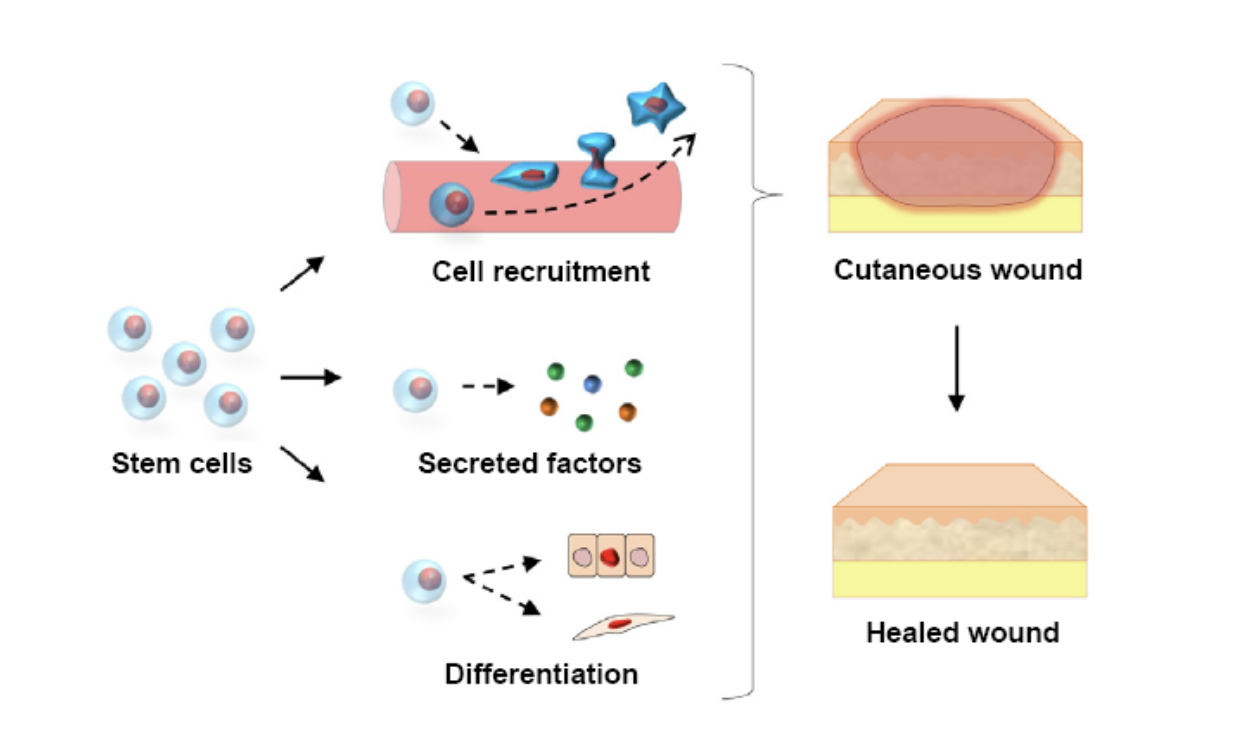
Acellular minimally manipulated tissue allografts are utilized to help treat a wide variety of conditions and are shown to help the body boost its ability to heal itself. The allografts help promote the body’s own healing process to assist in the reconstruction and regeneration of injured tissue. This can lead to an alleviation of pain, and a quick recovery.